Total solutions for every patient

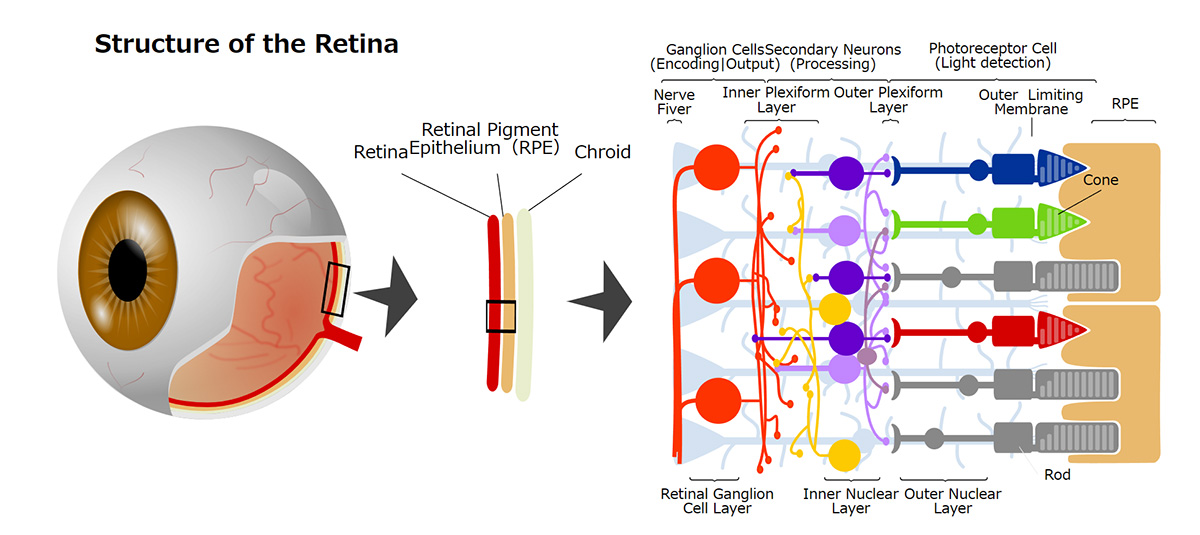
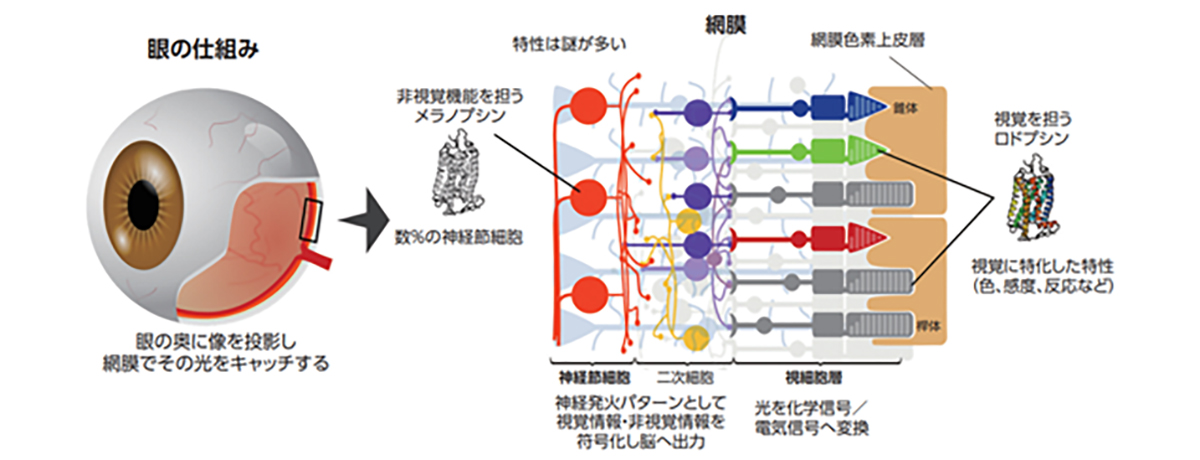
Photoreceptor cells are specialized nerve cells that act as light receptors, converting light into chemical signals and transmitting this visual information to secondary neurons, such as bipolar cells. These cells are located in the farthest layer of the retinal tissue within the eye, closest to the sclera (or the white part of the eye). They fall into two categories: rod cells, which facilitate vision in dim light, and cone cells, responsible for sharp vision and color perception. Cone cells are further divided into blue, green, and red types, based on their sensitivity to different light wavelengths, playing a crucial role in color vision. A distinguishing feature of photoreceptor cells is an intricate structure known as the outer segment, which efficiently captures light. This structure is densely packed with light-receptor molecules called opsins.
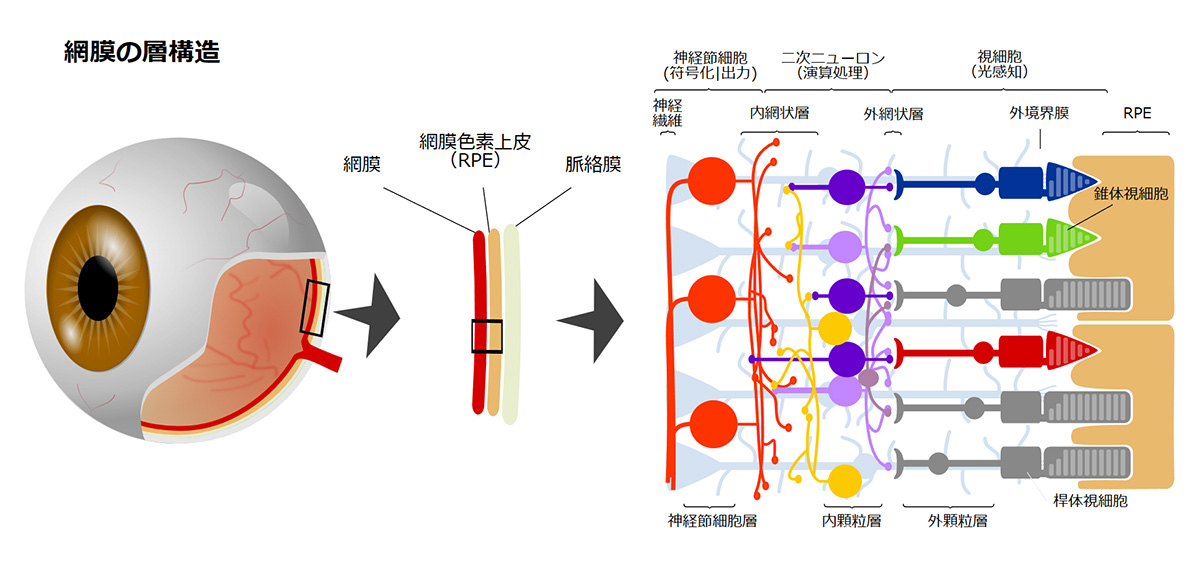
視細胞は光受容体として受け取った光を化学的シグナルに変換し、双極細胞などの二次ニューロンに光情報を伝える神経細胞で、視細胞は眼球内の網膜組織の最も外層(強膜側)に存在しており、薄明視を司る桿体視細胞と、視力や色覚を司る錐体視細胞の2種類のサブタイプに分類されます。錐体視細胞はさらに光の波長に対する感受性の違いから青、緑、赤錐体の3種類に分類され色覚をつかさどります。視細胞は外節と呼ばれる光を効率よく受け取るための特徴的な構造を有し、外節内には光受容分子であるオプシンが密に分布しています。
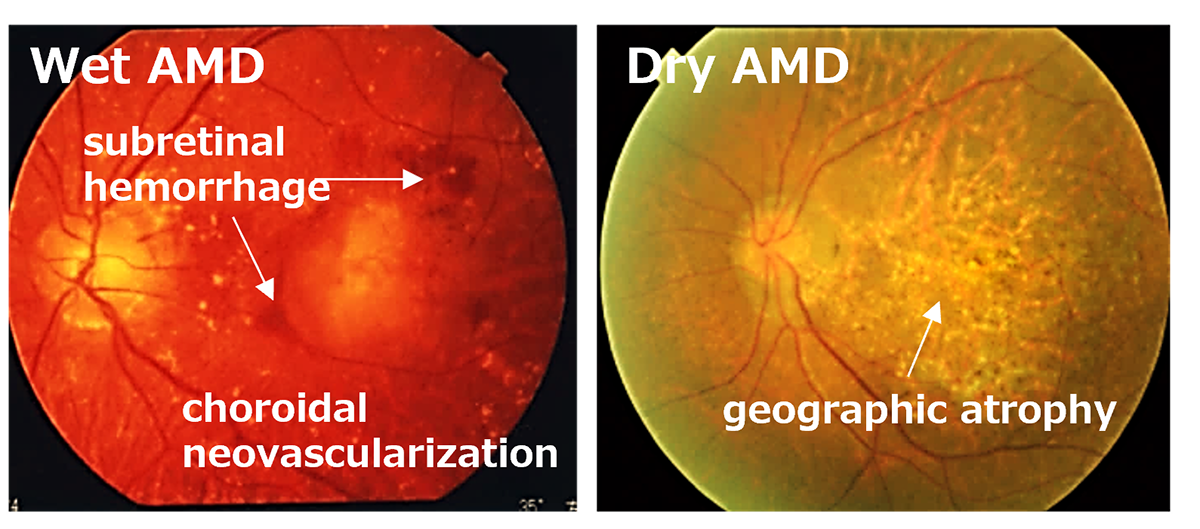
Age-Related Macular Degeneration (AMD) is one of the most common causes of blindness worldwide, particularly among the elderly. The global prevalence is 8.7% leading to an estimate that about 288 million people in western countries will be afflicted by 2040 (2). The early stages of AMD are characterized by features known as drusen and pigment loss from retinal pigment epithelium (RPE) cells. The progression of AMD is marked by the widespread degeneration of RPE cells and the formation of choroidal neovascularization (CNV). Advanced cases, directly linked to a decrease in quality of life (QOL) through central visual field impairment and visual impairment, are classified into two forms: atrophic and exudative (as shown below). In exudative forms, CNV leads to blood and lipid leakage beneath the retina, and fibrous scarring, which cause the degeneration of visual cells and visual impairment. Presently, the treatment consists of intravitreal injections of anti-vascular endothelial growth factor (anti-VEGF) agents for the exudative type, and complement inhibitory drugs for the atrophic type. These treatments aim to curb disease progression but require regular long-term intervention. However, neither treatment can restore visual function once the retina has already degenerated. Therefore, for the progressive cases of AMD, reconstructing the retina through cell transplantation remains the only means to preserve or restore visual function.
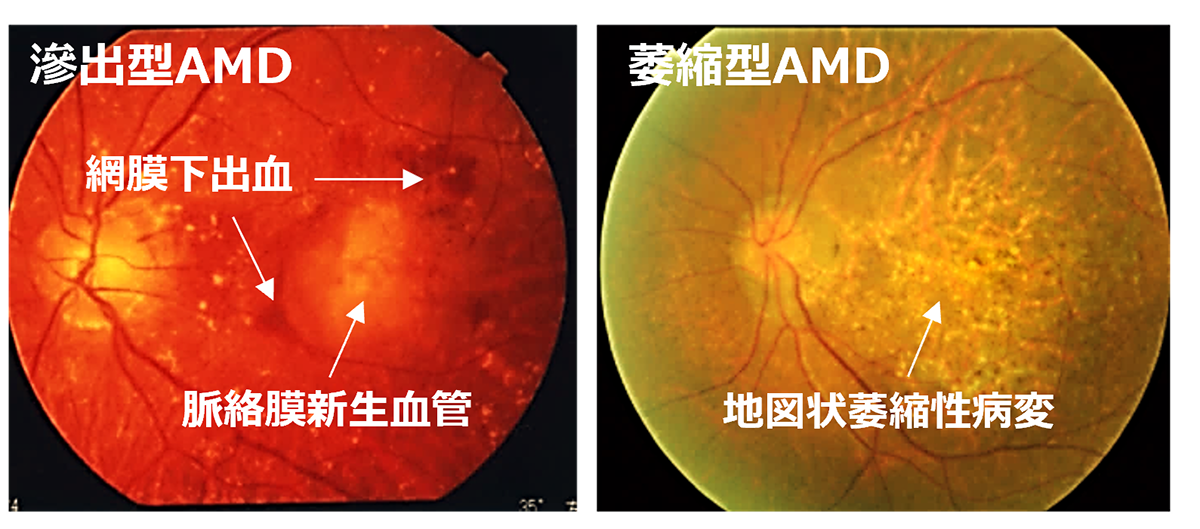
先進諸国における高齢者の主な失明原因の1つであるAMDの世界的な有病率は8.7%で、2040年までにグローバルで約2億8800万人が罹患すると予測されています。AMDの初期段階は、ドルーゼンおよびRPE細胞の色素脱失として認められ、進行するに従いRPEの広範囲な変性や脈絡膜血管新生(CNV)が形成されます。中心視野と視力障害というQOL低下に直結する視機能障害を特徴とする進行症例は、萎縮型と滲出型の2つの形態に分類されます。滲出型ではCNVが原因となり血液、脂質の網膜下漏出、および網膜下での線維性瘢痕の形成することで視細胞が変性し、視機能障害がおこります。現在、病状の進行抑制を目的とした対症療法として、滲出型に対して抗血管内皮成長因子(抗VEGF)剤、萎縮型に対して補体活性抑制薬の硝子体内注射が行われていますが、進行抑制のためには長期にわたる定期的な治療が必要となります。しかしながら、いずれの治療法においても既に変性を来した網膜の視機能回復は期待することはできません。そのため、AMDの進行症例に対する視覚機能を維持または回復するための根治治療には細胞移植による網膜再建が唯一の手段となります。
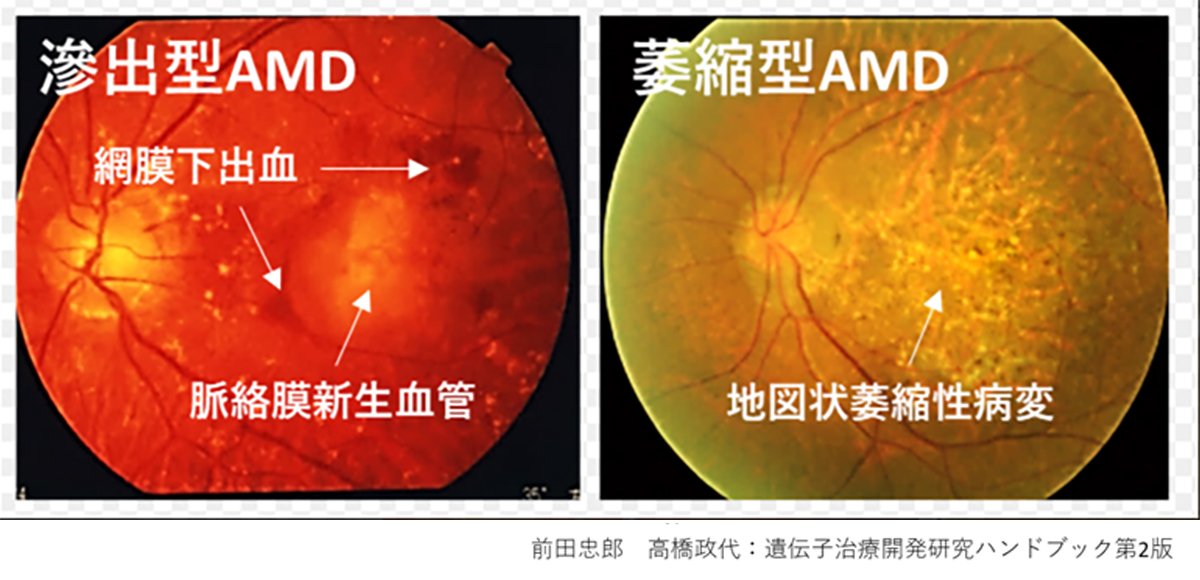
先進諸国における高齢者の主な失明原因の1つであるAMDの世界的な有病率は8.7%で、2040年までにグローバルで約2億8800万人が罹患すると予測されています(2)。AMDの初期段階は、ドルーゼンおよびRPE細胞の色素脱失として認められ、進行するに従いRPEの広範囲な変性や脈絡膜血管新生(CNV)が形成されます。中心視野と視力障害というQOL低下に直結する視機能障害を特徴とする進行症例は、萎縮型と滲出型の2つの形態に分類されます(下写真)。滲出型ではCNVが原因となり血液、脂質の網膜下漏出、および網膜下での線維性瘢痕の形成することで視細胞が変性し、視機能障害がおこります。現在、病状の進行抑制を目的とした対症療法として、滲出型に対して抗血管内皮成長因子(抗VEGF)剤、萎縮型に対して補体活性抑制薬の硝子体内注射が行われていますが、進行抑制のためには長期にわたる定期的な治療が必要となります。しかしながら、いずれの治療法においても既に変性を来した網膜の視機能回復は期待することはできません。そのため、AMDの進行症例に対する視覚機能を維持または回復するための根治治療には細胞移植による網膜再建が唯一の手段となります。
Initially, iPS cells were created from skin cells, but subsequent studies have shown that they can be generated from various types of cells. Nowadays, it’s more common to derive them from blood cells, making the process as simple as drawing blood. The genes used to induce iPS cells have also been modified, and the methods used to introduce these genes have evolved from retroviruses to plasmids and Sendai viruses. Today, iPS cells can even be created using mRNA. However, it’s important to note that iPS cells form a heterogeneous group, meaning they can greatly differ from each other. Even when generated using the same process, individual variations can lead to significant differences in gene expression levels. Moreover, the same cell line can behave differently depending on the cell culture techniques used. Even when following the same protocol, the cells can change differently during maintenance culture and passage, emphasizing the complexity of working with these cells.
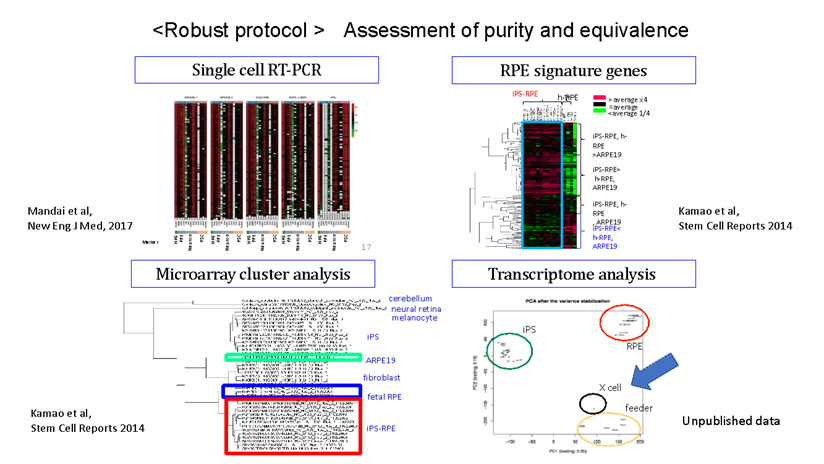
During our pionnering work with iPS cells, we leveraged insights from the broader field of stem cell research to guide the transformation of these iPS cells into Retinal Pigment Epithelial (RPE) cells. While iPS cells are heterogenous and exhibit significant differences even within the same line, when they are differentiated into RPE cells, these differences become insignificant. Historically, there are no instances of these RPE cells turning cancerous, making them a safe choice for our work. We devised a robust method that ensures the creation of consistently safe RPE cells with identical functionalities from each patient’s unique iPS cells(1). This strategic approach made it possible for autologous transplants, effectively bypassing potential issues of immune rejection.
Subsequently, utilizing the iPS cells created by the Center for iPS Cell Research and Application (CiRA) at Kyoto University that matched the 6-locus HLA of a patient, we derived RPE cells and successfully transplanted them into the retina of an elderly patient with age-related macular degeneration. Notably, this procedure was carried out without the use of systemic immunosuppressive drugs[2]. The findings suggest that, when there is a precise HLA match, potential rejection reactions can be efficiently controlled through local steroid administration alone.
Exploiting the insights gained from these clinical trials, we are currently developing treatments that could allow us to transplant cells into any patient without requiring immunosuppressive drugs. This involves creating RPE cells from iPS cells that have undergone genetic alterations to partially disable specific HLA genes, thereby creating a universal cell line applicable to all patients.
Reference:
(1)Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Kamao H, Mandai M, Okamoto S, Sakai N, Suga A, Sugita S, Kiryu J, Takahashi M. Stem Cell Reports. 2014 Jan 23;2(2):205-18.
(2)HLA-Matched Allogeneic iPS Cells-Derived RPE Transplantation for Macular Degeneration. Sugita S, Mandai M, Hirami Y, Takagi S, Maeda T, Fujihara M, Matsuzaki M, Yamamoto M, Iseki K, Hayashi N, Hono A, Fujino S, Koide N, Sakai N, Shibata Y, Terada M, Nishida M, Dohi H, Nomura M, Amano N, Sakaguchi H, Hara C, Maruyama K, Daimon T, Igeta M, Oda T, Shirono U, Tozaki M, Totani K, Sugiyama S, Nishida K, Kurimoto Y, Takahashi M. .J Clin Med. 2020 Jul 13;9(7):2217.
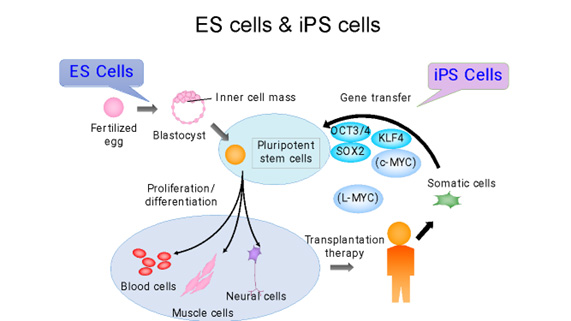
人工多能性幹細胞(iPS細胞)とは、成人の細胞のエピゲノムを初期化(リプログラミング)して胚性幹細胞(ES細胞)のような状態にした幹細胞の一種です。このプロセスにより、細胞は体内のほぼすべての種類の細胞に分化する能力を持つようになります。(図1)
iPS細胞は、2006年に京都大学の山中伸弥教授らによって初めて作られました。山中教授の研究チームは、まず、細胞の多能性(あらゆる種類の細胞になる能力)を維持する鍵になると思われる24の遺伝子を選び出し、成人の皮膚細胞に挿入しました。一つずつ遺伝子を減らす消去法で、これらの成体細胞を胚の状態に再プログラムするのに必要な遺伝子は4つだけであることを発見しました(1)。(それらはOct3/4、Sox2、c-Myc、Klf4であり、現在では山中因子として知られています。)この発見は、体内のあらゆる細胞を胚から得られる多能性幹細胞ES細胞様の細胞に変える可能性があることを意味し、タイムマシンの様な研究と呼ばれる革命的な発見でした。iPS細胞は患者自身の細胞から作ることができるため、移植による拒絶反応のリスクを軽減した個別化治療、創薬、病気のモデル化などに利用できる可能性があり、爆発的に研究が広がりました。その功績が認められ、細胞の初期化(リプログラミング)の概念に関する先駆的な研究を行ったジョン・ガードン博士と共に山中教授は「2012年・ノーベル生理学・医学賞」を受賞し、その後も数々の賞を受賞しました。
参考文献:
図1:
視細胞は光受容体として受け取った光を化学的シグナルに変換し、双極細胞などの二次ニューロンに光情報を伝える神経細胞で、視細胞は眼球内の網膜組織の最も外層(強膜側)に存在しており、薄明視を司る桿体視細胞と、視力や色覚を司る錐体視細胞の2種類のサブタイプに分類されます。錐体視細胞はさらに光の波長に対する感受性の違いから青、緑、赤錐体の3種類に分類され色覚をつかさどります。視細胞は外節と呼ばれる光を効率よく受け取るための特徴的な構造を有し、外節内には光受容分子であるオプシンが密に分布しています。
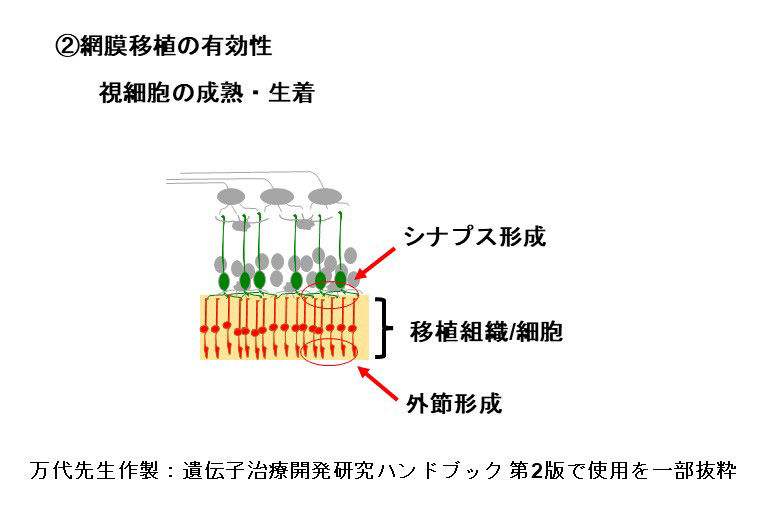
移植に用いる網膜オルガノイドは主に網膜前駆細胞と視細胞前駆細胞から構成されており、これらの細胞は移植後に成熟します。その結果、視細胞に外節構造が発達するとともにオプシンやトランスデューシンなどの機能分子も発現し、光報情伝達経路が発達します。また光情報を双極細胞を含む二次ニューロンに伝達するためのシナプスも形成されます。我々がヒト網膜オルガノイドから調整した網膜シートをモデル動物に移植したところ、移植視細胞は十分に成熟し、生体内の視細胞と同等の外節構造が発達することを電子顕微鏡を用いた観察で明らかにしました。光情報伝達に必要な機能分子が発現していることと、移植視細胞がホスト側双極細胞とシナプス結合していることを示唆する結果も免疫組織化学的手法により示しています。またサルを用いた移植試験から、移植後2年以上経過しても移植視細胞の生着状態が良好であることを確認しています。
移植視細胞がホスト網膜と機能的に繋がっていることを示すために、多電極電極アレイシステム(Multi-electrode-array, MEA)を用い電気生理学的に検証しました。具体的には視細胞が変性した末期の免疫不全網膜変性ラットにヒト網膜オルガノイドから調整した網膜シートを移植し生着させた後、ホスト網膜を移植網膜とともに取り出し、ホスト側神経節細胞の光に対する応答をMEAで記録しました。その結果、移植網膜が存在する部位でMEAの応答が検出されました。このことは移植視細胞が光に対して応答すること、さらにその信号がホスト網膜に伝達されていることを示唆しています。
私たちは移植により視機能の改善が見られるかを行動実験でも検証しています。マウスiPS細胞由来網膜オルガノイドを末期視細胞変性モデルマウスに移植後行った行動実験(shuttle aviodance test)では、網膜移植を行ったマウスが光に反応できたことから、光に対する移植視細胞の生理学的応答が行動に反映されることが示唆されました。また、人工的に視細胞変性を起こしたサルの網膜下にヒト網膜オルガノイドから調整した網膜シートを移植したケースでは、visually-guided saccades(VGS)テストにより視野の改善を示唆する結果が得られました。
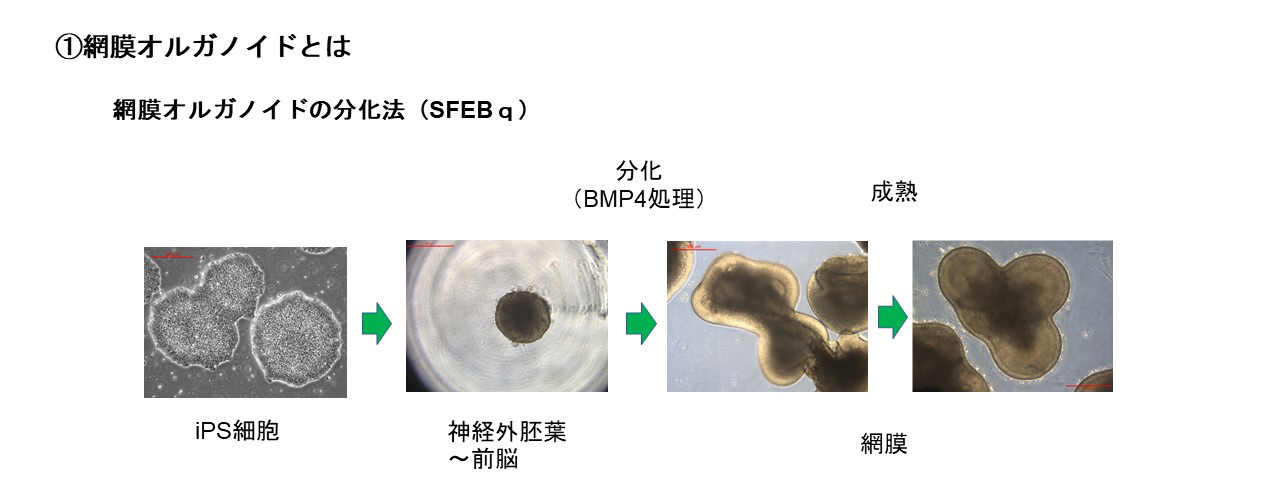
網膜オルガノイドとは生体内の網膜”様”の構造を持つ立体組織で、ES細胞(胚性幹細胞)やiPS細胞(多能性幹細胞)からin vitroで液性因子を用いて分化誘導することで作り出すことができます。発生過程はin vivoの網膜発生と同様に進行します。また組織形成は細胞間の相互作用のもと自律的に進行し、外顆粒層、内顆粒層、神経節細胞層の3層構造が形成されます。この過程において、視細胞を含む全ての細胞種も網膜前駆細胞から分化すします。
接着培養や浮遊培養などの培養方法や、誘導方法に用いる液性因子に多少の違いがありますが、ES/iPS細胞から神経外胚葉を経由し網膜に誘導する分化方法が一般的です。我々の開発ではTGFβファミリーに属するBMP4による網膜分化誘導方法を用いている。Serum-free Floating culture of Embryoid Body-like aggregates with quick reaggregation(SFEBq)法がその代表的手法で、ES/iPS細胞の凝集塊を浮遊培養し、BMP4を用いて網膜への分化誘導刺激を加えます。揺り戻し法を組み合わせることで非常に高品質な網膜オルガノイドを作製することが可能になります。実際、神戸アイセンター病院で2020年に行われた、網膜色素変性患者への世界初のiPS細胞由来網膜シート移植にはこの手法を用いて作製された網膜オルガノイドが用いられました。我々はこの誘導方法をベースに開発された、より大型の網膜オルガノイドの作製技術を用いて移植用網膜を作製しています。
網膜変性疾患の患者由来iPS細胞から網膜オルガノイドを分化した場合は、その病態の解析に用いたり、疾患の進行を抑制するのに有効な治療薬の開発などに用いることができます。我々は網膜オルガノイドを移植組織として用いることで、失った視野や視力の回復を目指しています。

